— Что именно называют клиническими исследованиями? Это весь процесс разработки лекарства или какая-то его часть?

Олег Розенсон
— Клинические исследования — это фаза, предшествующая регистрации и лицензированию лекарственного средства.
Сначала ведут поиск потенциальных молекул. Затем их эффективность и безопасность оценивают в лабораторных условиях на клеточных культурах или животных, что регулируется международными правилами. Если молекула показала положительные результаты на этапе доклинических исследований, препарат начинают оценивать в клинических исследованиях.
Клинические исследования до регистрации проходят три фазы. И в случае положительных результатов формируют досье для регистрации, которое подают в государственные органы.
Редко, но бывают ситуации, когда компания-разработчик считает результаты положительными, но государственные органы или наднациональные международные структуры — в России это Минздрав, в США — Food and Drug Administration (FDA), в Европейском союзе — European Medicines Agency (EMA) — признают данные недостаточными для регистрации лекарственного средства.
— Какой процент лекарств со стадии разработки доходит до пациента?
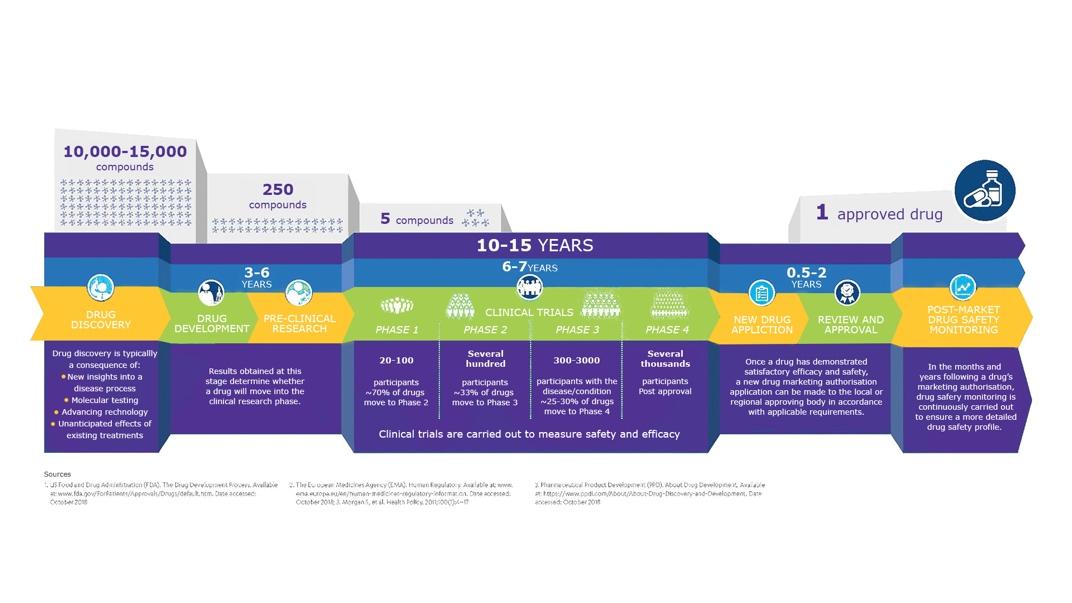
— Из 10–15 тысяч потенциальных молекул регистрируется всего одна. Поиск новых препаратов ведут, в первую очередь, исходя из нерешенных клинических вопросов. В последнее время при поиске и разработке новых лекарственных средств используют большие массивы данных и искусственный интеллект.
До стадии доклинических исследований из 10–15 тысяч молекул доходят в среднем 250. Доклинические исследования длятся 3–6 лет, и в среднем пять молекул из 250 выходят на клинические исследования.

Клинические исследования имеют четыре фазы. Первая фаза проходит на здоровых добровольцах. Ее цель — подтвердить безопасность препарата для человека, а также собрать данные по фармакокинетике. В среднем, она длится 1–2 года, в ней участвуют 20–80 пациентов.
Во второй фазе лекарственный препарат назначают уже пациентам с заболеванием, для лечения которого он разрабатывается. Вторая фаза занимает не менее 2–3 лет с участием 80–100 пациентов, и в ходе данной стадии оцениваются эффективность и безопасность, а также определяются минимальная эффективная доза и оптимальный режим дозирования.
В третьей фазе действие исследуемого препарата сравнивают со стандартным лечением. Она занимает 3–4 года и в ней участвуют 1000–4000 пациентов.
Четвертая фаза проводится после регистрации препарата и обычно ставит своей целью получение дополнительных данных как в контролируемых условиях, так и в условиях обычной клинической практики.
Из пяти лекарственных препаратов, дошедших до клинических исследований, регистрируется только один. Процесс регистрации занимает от 1,5 до 2 лет.
Таким образом, с момента поиска новой молекулы до его регистрации проходит 10–15 лет.
От трагических последствий — к единым правилам
— Как появились современные нормы проведения клинических исследований?
— В первую очередь, внимание уделялось этическим принципам проведения исследований. Правила формулировались в течение всего XX века — в основном, как результат трагических последствий отсутствия должного регулирования.
Первый такой пример — эликсир сульфаниламида, поступивший в продажу в 1937 году. Сам сульфаниламид — один из первых антибиотиков — успешно использовался и до этого, но производитель предложил новую удобную форму — сироп на основе диэтиленгликоля со вкусом малины. К сожалению, выбранный растворитель оказался чрезвычайно токсичным. Он вызывал острую печеночную и почечную недостаточность и привел к смерти первых же пациентов.
В результате уже в 1938 году конгресс США принял федеральный акт о пищевых продуктах, лекарственных средствах и косметике. Он предоставил государству контроль над разработкой лекарственных препаратов.
Следующим важным шагом стал Нюрнбергский кодекс 1947 года. Он стал ответом на бесчеловечные опыты на людях, которые проводились во время Второй мировой войны, и закрепил этические нормы обращения с пациентами в процессе клинических исследований.
В 1961 году была еще одна драматическая страница — вокруг уже испытанного лекарства — талидомида. Он обладал снотворным действием, и его начали прописывать беременным женщинам. Оказалось, что препарат был с тератогенным эффектом и приводил к врожденным уродствам.
Все эти печальные страницы истории привели к появлению в 1964 году Хельсинкской декларации Всемирной медицинской ассоциации (ВМА).
— Так как можно и как нельзя работать с испытуемым с точки зрения этических принципов?
— Основополагающим документом в вопросах этики медицинских экспериментов является Хельсинкская декларация. Она адресована врачам и призывает других лиц, вовлеченных в медицинские исследования с участием человека в качестве субъекта, следовать этим принципам. Последние изменения были внесены на 64-ой ассамблее ВМА в Форталезе в 2013 году.
Ниже приведены основные принципы Хельсинкской декларации:
- Основная цель клинических исследований (КИ) — понять причины, механизмы развития и последствия заболеваний; совершенствовать профилактические, диагностические и терапевтические вмешательства.
- Цель никогда не должна превалировать над правами и интересами отдельных субъектов исследования.
- Долг врача — защита жизни, здоровья, достоинства, неприкосновенности, права на самоопределение, частную жизнь и конфиденциальность персональных данных субъектов исследования.
- Адекватная этическая и научная подготовка, образование и квалификация лиц, проводящих исследование.
- Публикация не только положительных, но и отрицательных результатов, а также информация о проведенных исследованиях с неубедительными результатами.
- Соблюдение этических стандартов, гарантирующих уважение ко всем субъектам исследований и защиту их здоровья и прав.
- Привлекать пациентов только в той степени, в какой это оправдано с точки зрения потенциальной профилактической, диагностической или терапевтической ценности.
- Гарантия компенсации и лечения субъектам, пострадавшим в результате участия в КИ.
- Учет национальных, международных этических и правовых норм.
- Минимальный вред окружающей среде.
Польза от лечения должна быть выше рисков
— С какой целью проводятся клинические исследования?
— Клинические исследования проводятся, чтобы понять причины заболевания и механизмы его развития, а также совершенствовать его профилактику и терапию.
Но долг врача всегда — это защита здоровья и жизни человека и не только. Врач с должной этической и научной подготовкой понимает, что в зоне его ответственности также неприкосновенность личности пациента, его право в любой момент отказаться от лечения, его частная жизнь и конфиденциальность персональных данных.
Поэтому цель исследования никогда не может быть важнее прав и интересов его участников.
Пациентов привлекают только тогда, когда это оправдано с точки зрения их здоровья: риск не может быть выше, чем ожидаемая польза от лечения.
Специальные страховки гарантируют компенсацию и лечение участникам клинических исследований.
— А как понять, успешно ли исследование?
— Исследование конструируется таким образом, чтобы можно было сравнить предлагаемую терапию с наилучшей имеющейся на данный момент терапией или с плацебо, если таковой нет.
Также важно отметить, что современные нормы обязывают производителей публиковать все результаты исследований — не только положительные, но и отрицательные или нейтральные.
Важно, что с этической точки зрения исследование не должно проводиться повторно, если получены убедительные результаты. Тем самым минимизируются риски для пациентов, удается избежать «избыточного» числа исследований.
— Как регулируется проведения исследования?
— Для этого сегодня существует стандарт, который называется «Качественная клиническая практика» (GCP — Good Clinical Practice). Это международный стандарт, который позволил гармонизировать правила разных стран, то есть сделать их универсальными.
Это был большой шаг вперед — странам стало не нужно отдельно набирать свои когорты пациентов, причем произошло это без ущерба для качества исследования. Правила GCP были утверждены в ЕС и США в 1997 году, а в России принципы GCP были введены в 2005 году в ГОСТ Р 5279-2005 «Надлежащая клиническая практика».
Эти стандарты регулируют не только планирование и проведение исследований с участием человека, но и документальное оформление и представление результатов исследований. Последнее очень важно, потому что именно на основе этих документов государственные органы принимают решение о регистрации лекарственного препарата.
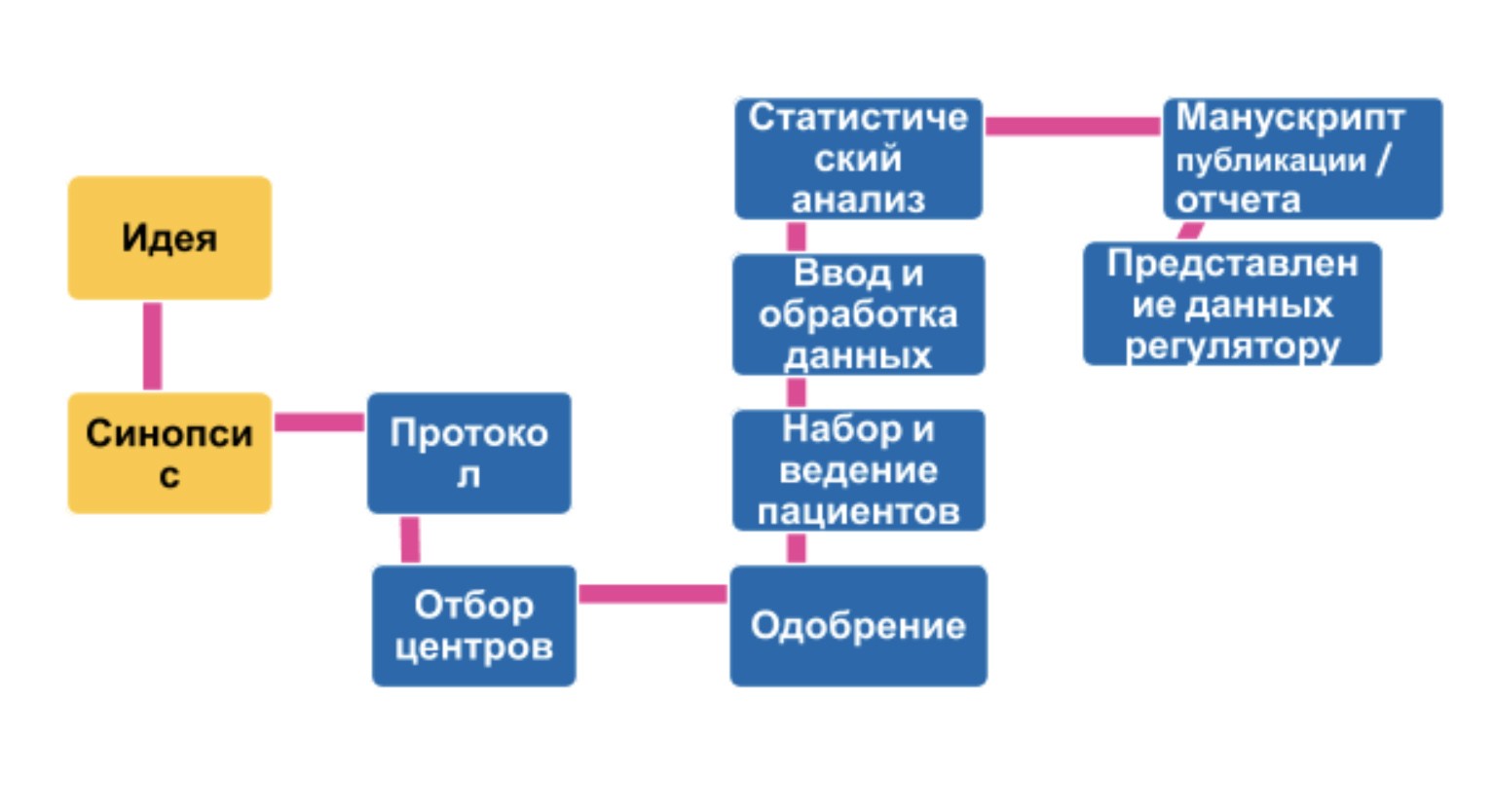
Синим помечены этапы, которые регулирует GCP стандарт
— Что гарантирует соблюдение стандарта «Качественная клиническая практика»?
— Соблюдение прав пациента и достоверность результатов исследований.
Это означает, что если после одобрения препарата в рамках обычной клинической практики доктора будут использовать его в соответствии с зарегистрированной инструкцией, то результаты будут сопоставимыми с результатами, полученными при клинических исследованиях.
— Как добиваются соблюдения этих правил?
— Любое клиническое исследование проходит две независимых экспертизы — научную и этическую.
Исследование рассматривает этическая комиссия. В ней должно быть не менее пяти человек, хотя бы один из них должен иметь квалификацию вне зоны медицины и еще один — не иметь отношения к организации, проводящей исследования.
Каждый пациент перед началом исследования подписывает форму информированного согласия, где подробно и понятно описаны цели исследования, права пациента, риски и ряд других стандартных пунктов. Важно, что пациент в любой момент может отозвать информированное согласие без объяснения причин. Тем самым подтверждается принцип добровольного участия любого пациента.